Document Type : Original Article
Authors
Department of Applied Chemistry, Faculty of Science, Malayer University, Malayer, 65174, Iran
Abstract
In this paper, by using of density function theory (DFT), we have investigated the interaction and adsorption of Cd+2 ion on the interior and exterior surface of pristine, C, P and C&P doped BNNTs. The calculated results indicate that the adsorption of Cd+2 is exothermic in thermodynamic approach. With adsorbing Cd+2 ion the electrical and optical properties of system alter significantly from original state. Inspection of quantum, natural bond orbital (NBO) and reduced density gradient (RDG) results confirm that the pristine and doped BNNTs are a good candidate to making sensor and adsorbent of Cd+2 in biological and environmental system.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Cadmium is a heavy metal and it is not a naturally occurring metal in biological systems. Cadmium physiologically exists as an ion (Cd+2) in the body. Cd+2 ion exposure is a risk factor associated with a large number of illnesses including kidney disease, early atherosclerosis, hypertension, and cardiovascular diseases. It can interact with different hormonal signaling pathways [1]. For this purpose, new research is focused to find a compound to detect and adsorbing Cd+2 in the environmental and biological system, one of them is nano material. After predicting boron nitride nanotube (BNNTs) by theoretical methods, it successfully synthesized in 1995 [2]. The behavior of BNNTs are very similar to carbon nanotubes (CNTs) and they are basically regarded as isomorphic analogs of the CNTs. The BNNTs have been favorable potential for applications in nanoelectronic devices for instance sensor and adsorbent of various gas molecules and other toxic materials [3‒6]. The chemical functionalization of BN nanotubes with various groups has been shown to be an efficient way to enhance their solubility and capability of nano tube to adsorb and sensor different materials [7‒12]. On the other hand, the doping and functionalizing impurity elements increase the reactivity and selectivity of the BNNTs toward molecular gas, HCN, CO, NO, NO2 [13‒17]. After our previous works on the study of interaction various materials with nanotube [18‒21], In the this project, DFT calculations are performed to investigate the interaction Cd+2 ion on the pristine, C, P and C&P-doped (4,4) armchair boron nitride nanotube. For this aims, at first step, the considered adsorption models of Cd+2 on the exterior and interior surface of BNNTs are optimized at the B3LYP/Lanl2DZ level of therory, and then, the electronic, structure properties, quantum parameters, adsorption energies, gaps energy, reduced density gradient (RDG), natural bond orbital (NBO) parameters for adsorption models are calculated and results are analysed.
Computational methods
The representative models of pristine and C, P, C&P doped BNNTs are individually optimized by using DFT theory at B3LYP/Lanl2DZ level of theory using the Gaussian 09 software [22].
Adsorption energy (Eads) of Cd+2 adsorption on the surface of BNNTs is calculated by:
Where EBNNTs-Cd+2 was obtained from the scan of the potential energy of the BNNTs–Cd+2, EBNNTs is the energy of the optimized BNNTs structure, and ECd+2 is the energy of an optimized Cd+2 and BSSE is base set superposition errors. The value of BSSE for all adsorption models is 0.002‒0.007 kcal/mol.
The quantum molecular descriptors electronic chemical potential (μ), global hardness (η), electrophilicity index (ω), energy gap, and electronegativity (χ) of the nanotubes [18‒21] are calculated as follows:
Where I (-EHOMO) is the ionization potential and A (-ELUMO) the electron affinity of the molecule.
Results and discussions
The electrical and structural properties
The interaction of Cd ion on the exterior and interior surface of pristine and C, P and C&P doped boron nitride nanotube is considered at the various configurations. The B and D models respectively denote the exterior and interior surface. The (I), (II), (III) and (IV) indexes are used for pristine, C, P and C&P doped BNNTs. All considered B‒I to D‒IV models are optimized.
The optimized structures of the B‒I to D‒IV models are shown in Fig .1. Inspection of optimized results reveal that the bond length (d) between Cd+2 and BNNTs is in range 1.30 to 1.95 Å. In all adsorption models, the shortest bond length is related to the (I) position and the longest bond length is related to the (III) position in Fig. 1.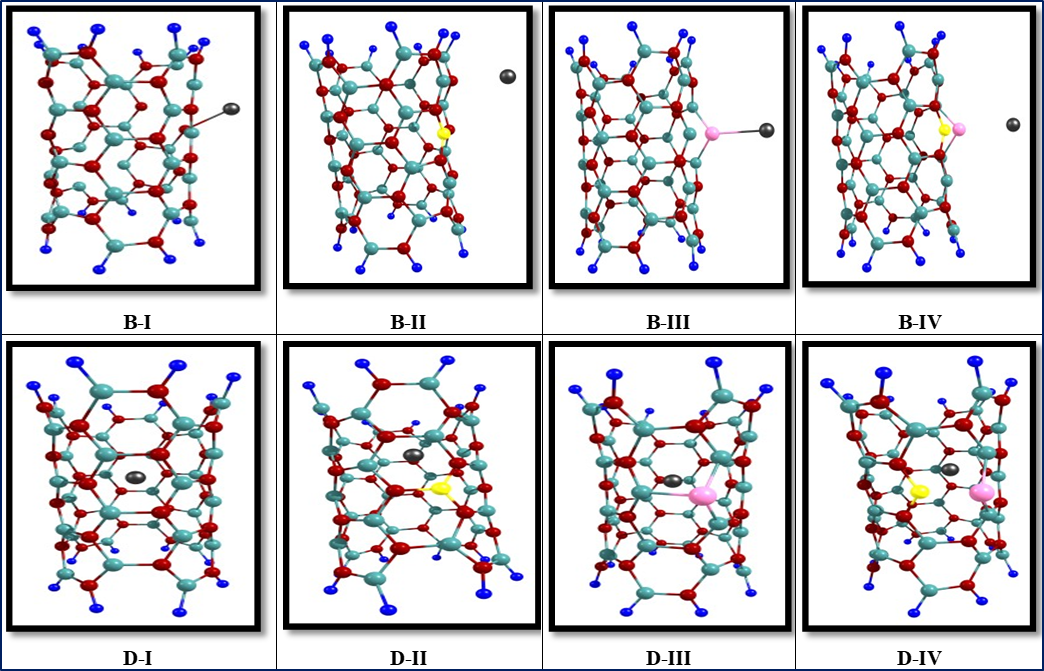
Fig. 1.
The adsorption energy of all adsorption models is calculated by Eq. 1, and calculated results are given in Table 1. As a shown, the adsorption energy of all adsorption models is in range ‒165.6 to ‒252.8 Kcal/mol and is exothermic in thermodynamic approach. These results confirm that the adsorption of Cd+2 on the surface of nanotube is chemisorption type. Comparison results indicate that with doping C, P and C&P atoms the amount of adsorption energy have been significantly reduced, and so the adsorption of Cd+2 on the exterior and interior surface of pristine model of BNNTs is more favorable than doped models. On the other hand, the adsorption of Cd+2 on the P doped models (B-III with ‒199.5 and D-III with ‒165.6 kcal/mol) are lower than C doped.
The amount of NBO charge transfer between nanotube and Cd+2 is in range ‒0.54 to ‒1.22 |e|. The negative value of charge transfer reveals that the Cd+2 has acceptor effect on the surface of nanotube and decrease the electron charge density around nanotube. The most values of charge transfer occurred in the D-IV model.
The HOMO and LUMO orbital descriptor
To understand the electrical properties of Cd+2 adsorption on the exterior and interior surface of BNNTs, the HOMO
(highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) and other quantum properties of system are calculated and results are given in Table 1 and are shown in Fig 2.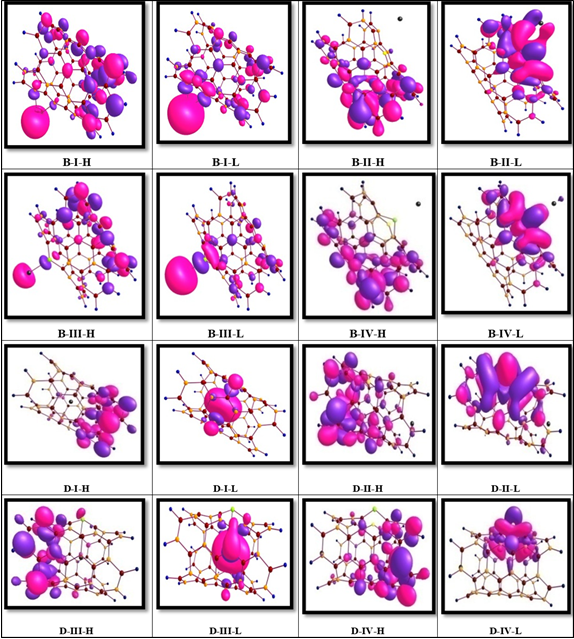
Fig. 2.
The eigenvalues of HOMO and LUMO and their gap energy reflect the reactivity, conductivity, and optical properties of the molecule. A molecule having small gap energy is more polarizable and is generally associated with a high chemical reactivity and low kinetic stability. The band gap energy of the pristine and C-doped BNNTs is 5.499, 3.68 eV respectively and agreement with other research [23‒24, 13]
The calculated results of HOMO-LUMO orbital density for all adsorption models reveal that the LUMO orbital density are localized around Cd+2 ion and on the adsorption area due to acceptor electron effect of Cd+2 ion. While the HOMO orbital density is widespread on the surface of the nanotubes. Therefore, the surface of the nanotubes is a good place to attack electrophilic species.
By using equations 2‒6 the quantum parameters such as electronic chemical potential (μ), global hardness (η), electrophilicity index (ω), electronegativity (χ) and gap energy (Egap) of the nanotubes are calculated and results are listed in Table 1.
Table 1. The adsorption energy (Kcal/mol), NBO charge and quantum parameters of Cd adsorption on pristine and C&Pdoped BNNTs
|
B-I |
B-II |
B-III |
B-IV |
D-I |
D-II |
D-III |
D-IV |
|||||
|
|
α |
β |
|
α |
β |
|
α |
β |
|
α |
β |
|
|
E(HOMO)/ev |
-11.5 |
-11.6 |
-11.63 |
-11.40 |
-11.4 |
-11.40 |
-12.10 |
-11.6 |
-11.6 |
-11.9 |
-11.4 |
-11.4 |
|
E(LUMO)/ev |
-11.01 |
-7.84 |
-10.61 |
-10.86 |
-8.14 |
-10.38 |
-10.64 |
-7.30 |
-10.02 |
-10.27 |
-7.59 |
-10.04 |
|
Egap/ev |
0.49 |
3.74 |
1.02 |
0.54 |
3.26 |
1.08 |
1.46 |
4.30 |
1.58 |
1.63 |
3.81 |
1.36 |
|
χ/ev |
11.29 |
9.76 |
11.12 |
11.12 |
9.79 |
10.88 |
6.41 |
9.4 |
10.8 |
11.15 |
9.5 |
10.7 |
|
η/ev |
0.24 |
1.87 |
0.51 |
0.27 |
1.63 |
0.54 |
0.73 |
2.15 |
0.79 |
1.63 |
1.90 |
0.68 |
|
ω/ev |
265.5 |
25.4 |
121.2 |
23.02 |
29.4 |
109.6 |
28.1 |
20.9 |
74.5 |
38.13 |
23.8 |
84.8 |
|
μ/ev |
-11.29 |
-9.76 |
-11.12 |
-11.15 |
-9.79 |
-10.88 |
-11.35 |
-9.40 |
-10.8 |
-11.1 |
-9.50 |
-10.7 |
|
Eads/Kcal/mol |
-452.8 |
-247.5 |
-199.5 |
-255.08 |
-426.8 |
-251.6 |
-165.6 |
-249.9 |
||||
|
Δρ (NBO) |
-1.18 |
-0.54 |
-1.18 |
-0.57 |
-1.10 |
-0.82 |
-1.10 |
-1.22 |
||||
Based on calculated results, HOMO and LUMO energies of the B‒I to D‒IV models are in range ‒11.4 to ‒12.1 and ‒7.3 to ‒11.01 eV respectively. With doping C atom in the B‒II, B‒IV, D‒II and D‒IV models the electron spin of nanotube is split to alpha and beta spin. The LUMO energy of alpha spin is decreased significantly from original values, whereas the HOMO energy of system is almost constant. Comparison results indicate that at the other models the HOMO and LUMO energies alter slightly from original values. The gap energy for B‒I to D‒IV models are in range 0.49 to 4.30 eV. The lowest value of gap energy occurred in the B‒I (0.49 eV) and the most value of gap energy occurred in D‒II alpha spin (4.30 eV). The lowest value of gap energy indicate the the most conductivity and reactivity of system. Inspection of results demonstrate that adsorbing Cd+2 decrease significantly the gap energy of system and so the conductivity and reactivity of system increase significantly from original state. This result confirms that the pristine and C, P and C&P doped BNNTs can be a good candidate to making a sensor and detector of Cd+2 ion for environmental and biological system.
The density of state (DOS) plots for all adsorption models are calculated in interval ‒20 to 5 eV by using Gaussum software and results are shown in Fig. 3. The DOS results show that in the HOMO and LUMO region it can be seen 14 and 12 maximum peaks respectively. With doping of C, P and C&P atoms and adsorbing the Cd+2 ions, the number of maximum peaks in the HOMO and LUMO area has not changed, but the height of the maximum peaks in these two regions has decreased significantly.
As we know, the global hardness is the resistance chemical systems towards the deformation of electron cloud under small perturbation encountered during the chemical process. According to calculated results of Table 1, the global hardness of the B‒I to D‒IV adsorption models change from 0.24 to 2.15 eV. With doping C tom the global hardness of alpha spin of B and D models increase significantly from original state and so the activity of system decrease significantly from original state.
The chemical potential of system is in range ‒9.5 to ‒11.35 eV and it demonstrates that with doping C atom and Cd+2 adsorbing the stability of system similar global hardness change slightly from original state. The electrophilicity index of system is in range 23.8 to 265.5 eV and reflects the conductivity of system. Therefore, in the B‒I model the conductivity of system is bigger than other models due to bigger value of electrophilicity index.
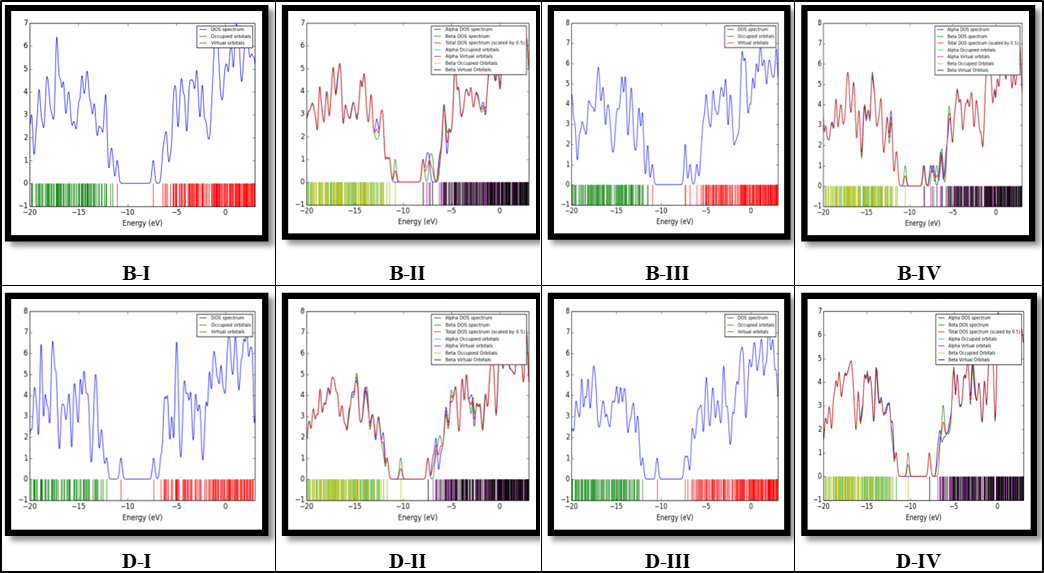
Fig. 3.
Natural bond orbital analysis
The natural bond orbital (NBO) is used to explore the charge transfer or conjugative interaction in molecular systems and is an efficient method for studying intra and intermolecular bonding and interaction among bonds. From NBO analysis, we determine the donor and acceptor orbital and the stabilization energy (E2) associated with the delocalization i→j as follow:
Where qi is donor orbital occupancy, and are orbital energies and Fij is the diagonal NBO Fock matrix element. The larger values of E(2) indicate, the strong interaction between electron donors and electron acceptors, and the more donating tendency from electron donors to electron acceptors and the greater the extent of conjugation of the whole system.
The results of E(2) value for all adsorption models around doping position are given in Table 2.
Table 2. The donor and acceptor orbital for Cd adsorption on pristine and C&P doped BNNTs
|
Donor |
Acceptor |
E(2)/ kcal/mol |
Ei-Ej (a.u) |
F(i ,j) (a.u) |
Donor |
Acceptor |
E(2)/ kcal/mol |
Ei-Ej (a.u) |
F(i, j) (a.u) |
||||
|
B-I |
σВ42-Ν41→ |
σ*B52-Ν52 |
3.16 |
|
1.06 |
0.05 |
D-I |
σВ42-Ν41→ |
σ*B52-Ν52 |
3.03 |
1.08 |
0.05 |
|
|
σВ42-Ν41→ |
σ*B51-Ν51 |
3.39 |
|
1.04 |
0.05 |
σВ42-Ν41→ |
σ*B51-Ν51 |
2.65 |
1.07 |
0.04 |
|||
|
σВ42-Ν41→ |
σ*B32-Ν32 |
3.25 |
|
1.06 |
0.05 |
σВ42-Ν41→ |
σ*B32-Ν32 |
3.11 |
1.08 |
0.05 |
|||
|
σВ42-Ν41→ |
π*B31-Ν31 |
3.48 |
|
0.34 |
0.1 |
σВ42-Ν41→ |
σ*B31-Ν31 |
3.7 |
1.07 |
0.05 |
|||
|
B-II |
σВ42-Ν41→ |
σ*B52-Ν52 |
2.14 |
|
1.21 |
0.06 |
D-II |
σВ42-Ν41→ |
σ*B52-Ν52 |
2.02 |
1.21 |
0.02 |
|
|
σВ42-Ν41→ |
σ*B51-Ν51 |
0.53 |
|
1.29 |
0.03 |
σВ42-Ν41→ |
σ*B51-Ν51 |
0.55 |
1.28 |
0.03 |
|||
|
σВ42-Ν41→ |
σ*B32-Ν32 |
2.01 |
|
1.21 |
0.06 |
σВ42-Ν41→ |
σ*B32-Ν32 |
2.16 |
1.21 |
0.06 |
|||
|
σВ42-Ν41→ |
σ*B31-Ν31 |
0.68 |
|
1.27 |
0.03 |
σВ42-Ν41→ |
σ*B31-Ν31 |
0.52 |
1.29 |
0.03 |
|||
|
B-III |
σВ42-Ν41→ |
σ*B52-Ν52 |
4.77 |
|
0.86 |
0.05 |
D-III |
σВ42-Ν41→ |
σ*B52-Ν52 |
4.93 |
0.81 |
0.05 |
|
|
σВ42-Ν41→ |
σ*B51-Ν51 |
3.15 |
|
0.87 |
0.04 |
σВ42-Ν41→ |
σ*B51-Ν51 |
3.86 |
0.78 |
0.05 |
|||
|
σВ42-Ν41→ |
σ*B32-Ν32 |
5.01 |
|
0.86 |
0.05 |
σВ42-Ν41→ |
σ*B32-Ν32 |
4.70 |
0.79 |
0.05 |
|||
|
σВ42-Ν41→ |
σ*B31-Ν31 |
2.96 |
|
0.87 |
0.04 |
σВ42-Ν41→ |
σ*B31-Ν31 |
3.47 |
0.78 |
0.04 |
|||
|
B-IV |
σВ42-Ν41→ |
σ*B52-Ν52 |
3.68 |
|
0.91 |
0.07 |
D-IV |
σВ42-Ν41→ |
σ*B52-Ν52 |
3.47 |
0.9 |
0.07 |
|
|
σВ42-Ν41→ |
σ*B51-Ν51 |
0.88 |
|
0.98 |
0.03 |
σВ42-Ν41→ |
σ*B51-Ν51 |
1.21 |
0.95 |
0.04 |
|||
|
σВ42-Ν41→ |
σ*B32-Ν32 |
3.82 |
|
0.91 |
0.07 |
σВ42-Ν41→ |
σ*B32-Ν32 |
3.38 |
0.9 |
0.07 |
|||
|
σВ42-Ν41→ |
σ*B31-Ν31 |
0.85 |
|
0.98 |
0.03 |
σВ42-Ν41→ |
σ*B31-Ν31 |
1.12 |
0.95 |
0.04 |
|||
The strong intramolecular hyper conjugative interaction of donor orbital to acceptor orbital for A‒a to B‒e models occur in the and . The lowest value for E(2) in the all models occur in the . It is notable that with doping C atom in all adsorption models the E(2) value decrease significantly from original value and so the lowest charge transfer is occurred in them. These facts may be the probable reasons behind the relative instability of the axial and equatorial adsorption C atom doped on the outer surface of BNNTs based on energetic data and NBO interpretation.
Reduced density gradient (RDG) and NCI index
To understand the intramolecular interactions and evaluate the nature of the weak interactions, the non‒covalent interaction index (NCI) for the complexes considered have been calculated. The NCI index provides more evidence related to the non‒covalent interaction. The reduced density gradient (RDG) is defined [25]:
Non‒covalent interactions are characterized by small values of RDG. These isosurface expand over interacting regions of the complex. The product between electron density ρ(r) and the sign of the second lowest eigenvalues of electron density hessian matrix (λ2) has been proposed as a tool to distinguish the different types of interactions. The scatter graphs of RDG versus sign(λ2) ρ for all adsorption models are shown in Fig. 4.
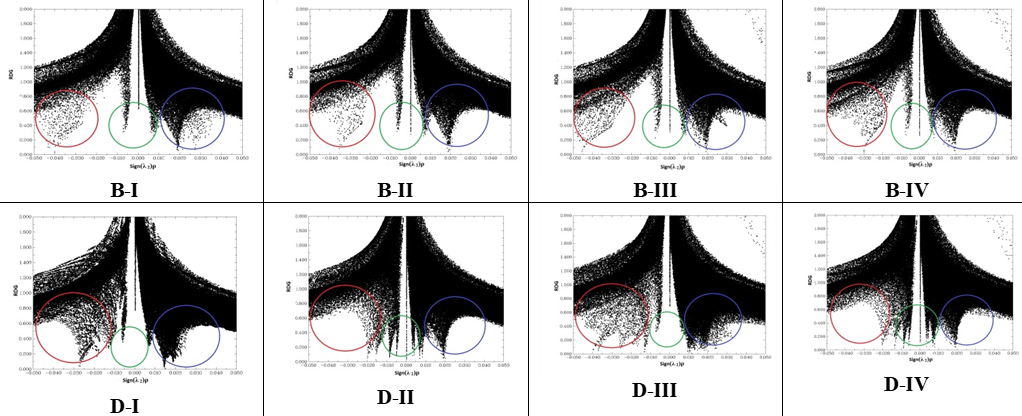
Fig. 4.
The X‒axis and Y axis are sign(λ2)ρ and RDG function respectively. The sign(λ2)ρ(r) and NCI‒RDG plots are obtained with Multiwfn program [26]. In the RDG scatter graph, red color circle shows the attractive interactions, blue color circle denotes strong repulsive interactions and green circle implies low electron density, corresponding to Van der walls interactions. It clearly observed that in the all adsorption models except D‒II and D‒IV the most electron density localized in λ2
Conclusions
In the current work, the interaction of Cd+2 on the surface of pristine and C, P and C&P doped BNNTs is investigated by using density function theory. The adsorption energy values of the all adsorption models are negative, and exothermic in thermodynamic approach. The RDG results demonstrate that the adsorption of Cd+2 is strong electrostatic. The NBO and quantum parameters reveal that with adsorbing Cd+2 the electrical, optical and conductivity properties of nanotube increase significantly from original state. These results demonstrate that BNNTs is a good candidate to making sensor, detector, and adsorbent for Cd+2 in environmental and biological system.
Acknowledgments
KSR thanks the Rural Development society (RDS) and DST, New Delhi, India, for financial support.
)